Pulmonary Hypertension WHO Classification
Pulmonary hypertension (PH) is a severe, rare lung disease characterized by high blood pressure in the pulmonary arteries. Due to the condition, the pulmonary arteries, which are responsible for transporting the blood from the right heart ventricle to the lungs, become narrowed and thickened. In order to properly pump the blood, the heart needs to work harder, which can lead to enlargement and weakening of the organ, as well as potential right heart failure.
There is currently no cure for pulmonary hypertension, but there are treatments that can help patients cope with the symptoms, which include shortness of breath, tiredness, chest pain or pressure, irregular heartbeat, pain on the upper right side of the abdomen and decreased appetite. Symptoms, diagnosis and treatment of pulmonary hypertension are different according to the different subtypes of the disease. In order to produce, maintain and implement international health information standards, the World Health Organization (WHO) defined a PH classification.
How Is The Pulmonary Hypertension WHO Classification Determined
The International Classification of Diseases (ICD) is created by the organization and comprises a standard diagnosis tool for physicians, nurses, other providers, researchers, health information managers and coders, health information technology workers, policy-makers, insurers and patient organizations. To determine the classification of pulmonary hypertension, the WHO analyzed the general health situation of patients, as well as the disease’s incidence and prevalence.
The pulmonary hypertension WHO classification is used for numerous purposes, including records, death certificates, information storage, diagnosis retrieval, national mortality and morbidity statistics, reimbursement, resource allocation, and other clinical, epidemiological and quality purposes, and the organization expects it to help fight the disease.
Different Pulmonary Hypertension WHO Classification Groups
The pulmonary hypertension WHO classification was updated in 2009, placing a greater importance on treating the underlying cause of the disease. There are now five groups, while the previous classification was more focused on whether the underlying cause was known and included only two categories: primary or idiopathic pulmonary hypertension, and secondary pulmonary hypertension. In all of the different groups, the average pressure in the pulmonary arteries is 25 mmHg or more at rest or 30 mmHg during physical activity, the normal levels between 8 and 20 mmHg at rest.
- Group 1 includes the subtypes of pulmonary hypertension associated with abnormalities in the arterioles, the small branches of the pulmonary arteries, such as pulmonary arterial hypertension (PAH), and idiopathic pulmonary hypertension (IPH), which is the type of PH diagnosed when the causes of the disease are not known. Also in this group is inherited pulmonary hypertension, as well as pulmonary hypertension caused by connective tissue diseases that affect the body’s structure or composition of the tissue like scleroderma, congenital heart problems, high blood pressure in liver (portal hypertension), HIV, thyroid gland disorder, sickle cell disease, glycogen storage disorders, and rare blood conditions like pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis.
- Group 2 refers to pulmonary hypertension caused by left heart disease, including cardiomyopathy, diastolic dysfunction, mitral stenosis, mitral regurgitation, aortic stenosis, and aortic regurgitation. The left side of the heart pumps blood to the entire body but the lungs, but about 60% of patients with severe left ventricle dysfunction also develop pulmonary hypertension, since the blood flows through the entire heart, affecting the right side as well.
- Group 3 includes pulmonary hypertension resulting from lung diseases or shortage of oxygen in the body (hypoxia), with chronic obstructive pulmonary disease (COPD); interstitial lung disease; and sleep-disordered breathing, a group of diseases that affect the breathing during the sleep like obstructive sleep apnoea (OSA) being the most common diseases that can result in pulmonary hypertension.
- Group 4 refers to pulmonary hypertension secondary to blood clots, which are the body’s response to bleeding and injuries but can harm the heart and lungs when they occur without an apparent cause. Pulmonary embolus are blood clots that travel to the lungs and pulmonary thrombosis are clots that are formed in the lungs, which can block the pulmonary arteries.
- Group 5 is the last category and it includes other less common causes that do not fit into any of the other four groups. Sarcoidosis, which is a condition that results in inflammation of different organs like the lungs and lymph nodes, histiocytosis X, a rare disorder that causes scarring, granulomas and air-filled cysts mostly in the lungs, as well as compression of the blood vessels in the lungs, which can occur for numerous reasons like tumors, are some of the causes for pulmonary hypertension in the fifth group.
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) involves abnormally high blood pressure (hypertension) in the pulmonary artery. The pulmonary artery carries blood from the heart (right ventricle) to the lungs. When the smallest of these vessels become narrower in diameter (perhaps due to disease), this increases resistance to blood flow through these vessels. This increases the pressure inside these vessels as well as the right ventricle. Through time, the right ventricle has to pump harder and harder to move blood into the pulmonary artery, causing the heart to become enlarged and weakened. Eventually, the heart (right ventricle) becomes so weakened that it can no longer pump blood and heart failure occurs.
Other names for PAH include Ayerza syndrome, Familial (heritable) primary pulmonary hypertension (FPPH), Idiopathic pulmonary hypertension (arterial), Primary pulmonary hypertension and Sporadic primary pulmonary hypertension.
Pulmonary Arterial Hypertension Causes
As there are many causes of Pulmonary Arterial Hypertension, we’ll keep the list general:
- Any number of heart diseases can generate PAH
- Viral infections such as HIV
- Genetic mutations (this is known as familial or heritable PAH)
- Drugs such as cocaine and amphetamines and some prescription diet medications
- Scleroderma (autoimmune disease) which is a connective tissue disease that causes fibrosis
- Chronic liver diseases
- Unknown cause (can’t find a cause) which is referred to as idiopathic PAH
Pulmonary Arterial Hypertension Symptoms
Symptoms of PAH present when the increased blood pressure can’t fully overcome the increased resistance of the pulmonary arteries. This not only has effects on the heart and lungs but to the entire body.
In early phase PAH, patients may experience shortness of breath known as dyspnea during exercise along with possible fainting. As the disease progresses, these symptoms will occur even when patients are at rest:
- Dizziness
- Swelling of the legs and ankles known as edema.
- Pain in the chest region.
- tachycardia (fast heart rate) and irregular heart beat (palpitations or arrhythmias).
Pulmonary Arterial Hypertension Diagnosis
A doctor will ask potential PAH patients about family and medical history, and perform a physical exam and determine symptoms. From there s/he will try to confirm PH with tests, such as an echocardiogram, chest X-ray, electrocardiogram (EKG) and/or right heart catheterization. To try and determine the underlying cause, a doctor may perform Lung function tests, chest CT and MRI scans, and/or Polysomnograms. Please refer to the introduction on PH for an explanation of these various procedures.
Pulmonary Arterial Hypertension Treatments
Currently, there is no cure for any of the pulmonary hypertension types, however there are treatments that can help mitigate with symptoms. What therapies or combination of therapies are used depend on the underlying cause(s). The goals of such treatments ares to:
- Treat the underlying cause if it is known. It is reported that some therapies can correct some forms of PAH in individuals with secondary pulmonary arterial hypertension (SPAH).
- Improve the quality of life by reducing symptoms.
- Reduce the heart’s workload by increasing the supply of blood and oxygen.
- Reduce the production of blood clots and slow down the proliferation of smooth muscle cells in the blood vessel walls.
Treatments that are available include prescription drugs, oxygen therapy, lung transplants and possibly stem cell therapy.
Pulmonary Arterial Hypertension Medications
- Blood thinners are commonly used to decrease the production of blood clots.
- Calcium channel blockers help to relax blood vessels. This allows for an increase in blood flow and oxygen to the heart.
- Prostacyclin is administered to relax arteries in the lungs and prevents blood clots from forming. There are several forms of this medication. Newer forms of prostacyclin can be inhaled through a nebulizer for fast symptomatic relief.
- Nitric oxide, which can be inhaled has the ability to relax pulmonary arteries.
- Diuretics which help to remove excess fluid from the body reducing edema.
- Phosphodiesterase 5 inhibitors (PDE5) allows blood vessels to relax in the lungs.
Oxygen Therapy
As PAH progresses, blood oxygen levels decline, so patients may need oxygen therapy. The oxygen is administered through a mask or nasal prongs.
Lung Transplantation
If medical treatments are no longer an option for a PAH patient, they may require surgery to replace the diseased lung(s). A donor is required. Patients will have to take medication for the rest of their life to reduce the risk of rejection.
Pulmonary Arterial Hypertension Prognosis
Progress is being made to bring about new diagnostic tools and more efficient medications, however PAH is a fatal disease. It is reported that the survival rate is around 50 percent after 5 years.
Note: Pulmonary Hypertension News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Pulmonary Venous Hypertension
Pulmonary venous hypertension (PVH) differs from PAH in that high blood pressure occurs when the heart can’t efficiently carry blood away from the lungs. This results in blood collecting in lung tissue. This is generally caused by diseases of the left side of the heart. One such disease is endomyocardial fibrosis. Fibrous connective tissue can accumulate in one or both ventricles and results in a constricted (narrowed) heart chamber. This may also involve other heart structures such as the heart valves causing them to open and close abnormally.
Pulmonary Venous Hypertension Symptoms
PVH presents with many of the same symptoms seen in other forms of HP, however, it differs from PAH in that PVH’s shortness of breath occurs when a patient is in a reclined position or when sleeping. Symptoms include:
- Increased blood pressure
- Fluid in the lungs and between the layers that surround the lungs and chest wall
- Night breathing difficulty
- Fatigue
- Edema
- Decrease in urine production
- Coughing that may produce bloody mucus
- Decrease in overall heart function
Pulmonary Venous Hypertension Diagnosis
Your healthcare provider will want to know about your medical and family history. A physical exam will be administered to look for known symptoms of PVH such as a loud S2 (valve closure), jugular vein distention (jugular vein protrudes from the side of the neck), foot swelling and abdominal swelling (ascites).
Pulmonary Venous Hypertension Treatment
The goal is to try and treat the underlying causes. To prevent congestive heart failure, medications such as beta blockers, ACE inhibitors and diuretics are used. If a patient has structural heart issues, they may need surgical intervention.
Beta blockers block the effects of the sympathetic innervation of the heart. This decreases the hearts workload, so it needs less blood and oxygen. This aids in lowering blood pressure and helps to control abnormal beat rhythms that are either too fast or irregular. ACE (angiotensin-converting enzyme) inhibitors block the enzyme that is needed to form a molecule that constricts blood vessels. This allows blood vessels to relax allowing for easier flow of blood while reducing blood pressure. Diuretics help to remove excess fluid from the body decreasing edema (swelling).
Pulmonary Venous Hypertension Prognosis
PVH survival rates are essentially no different from other forms of PH. Depending on early diagnosis (if possible) and various therapies, an individual may survive somewhere between 3 to 10 years.
Pulmonary Hypoxic Hypertension
Pulmonary hypoxic hypertension as the name suggests involves high blood pressure and low oxygen (hypoxic) levels. This results from constricted pulmonary arteries to certain areas of the lungs. This phenomenon may seem illogical in that the pulmonary arteries actually constrict in the presence of low oxygen levels and this occurs without high carbon dioxide levels. Many would think that the arteries would relax and allow for greater blood flow to get more oxygen exchange. Under normal conditions, this is a mechanism to redirect blood flow to alveoli that have higher oxygen content. This constriction changes the distribution of blood flow in the lungs to areas of the lung that have more air. This increases the total area available in gas exchange since blood is not flowing to poorly ventilated aveoli. However, in the case of long-term whole-body hypoxia such as in HP, this works against the system. This is often seen in COPD and heart failure. Going to high altitudes can bring about the same phenomenon.
Pulmonary hypoxic hypertension Symptoms
- Pulmonary hypoxic hypertension shares many of the same symptoms as other forms of HP.
- Asymptomatic initially
- Pain in the chest
- Fainting spells and dizziness
- Shortness of breath
- Fatigue
- Edema of the feet and ankles (fluid retention and swelling)
- Jugular vein distention
- Ascites (swollen abdomen due to fluid buildup)
- Clubbing of fingers
- Increased blood pressure in the vessels leading to the lungs
Pulmonary Hypoxic Hypertension Diagnosis
For a PHH diagnosis, a doctor will explore medical and family history. A physical exam will be done to determine what symptoms a patient has. To confirm that a person has some form of PH, certain tests will be run, such as an echocardiogram, chest X-ray, electrocardiogram (EKG) and/or right heart catheterization. If it is determined that a person has pulmonary hypoxic hypertension, a doctor will try to determine the underlying cause. To do this s/he will run Lung function tests, chest CT and MRI scans, and/or Polysomnograms.
Pulmonary Hypoxic Hypertension Treatments
Treatments for Pulmonary hypoxic hypertension are pretty much the same as for other forms of HP which include:
- Diuretics to reduce fluid buildup in the body (edema)
- Blood thinners (anticoagulants) to prevent clot formation
- Digoxin which is used to treat congestive heart failure. This slows down heart rate during atrial fibrillation (a rapid, irregular heart beat).
- Oxygen therapy
- Calcium channel blockers for vessel relaxation
- Prostacylins for blood vessel relaxation and to prevent clot formation
- Heart or lung transplant
- Medications to treat the underlying cause if known
Pulmonary Hypoxic Hypertension Prognosis
Early diagnosis is important as well as finding the appropriate therapy(s), however there is no cure.
Thromboembolic Pulmonary Hypertension
When thromboembolic pulmonary hypertension becomes chronic, it causes severe pulmonary hypertension. Unfortunately, this form of HP goes under diagnosed. The lesions involve clots (thrombi) within the lumen of the blood vessels and fibrous stenosis (blood vessel constriction) or total destruction of the pulmonary arteries. As a result, the pulmonary vascular resistance increases generating pulmonary hypertension and progressive right heart (right ventricle) failure. Current research suggests that this disease is initiated by pulmonary embolism (as a single or recurrent events) and followed by progressive pulmonary vessel remodeling (angiogenesis).
Thromboembolic Pulmonary Hypertension Causes
Although there can many causes of thromboembolic pulmonary hypertension, patients are at high risk if they have experienced recurrent pulmonary thromboendartectomy (PTE), splenectomy (spleen removal) or lupus anticoagulant syndrome (antiphospholipid antibody syndrome). PTE is an operation that removes blood clots from the pulmonary arteries. Lupus anticoagulant syndrome causes blood clot formation in blood vessels and is an autoimmune disease that produces antibodies against a particular phospholipid known as cardiolipin and β2 glycoprotein.
Thromboembolic Pulmonary Hypertension Symptoms
The symptoms of thromboembolic pulmonary hypertension are essentially the same as other forms of pulmonary hypertension which include:
- Progressive breathing difficulty upon exercise
- Hemotysis which means coughing and expulsion of bloody mucus
- Fatigue
- Fainting spells and dizziness
- Syncope (brief loss of consciousness due to lack of oxygen)
- Edema which is fluid retention in the body
- Embolism due to clot formation (causes blood vessel obstruction)
- Systolic murmur of the tricuspid valve (regurgitation)
- Extended neck veins
- Ascites (swollen abdomen from fluid build up)
- Cyanosis such as blue lips and skin due to lack of oxygen
Thromboembolic Pulmonary Hypertension Diagnosis
Treatments for thromboembolic pulmonary hypertension is considerable different from PAH so it is of the essence to determine that the patient has thromboembolic type. One of the difficulties is that many patients have no history of pulmonary embolism which makes it difficult to diagnose properly. Some of these patients will be misdiagnosed.
Tests to Confirm Thromboembolic Pulmonary Hypertension
- Echocardiography – This is the first diagnostic tool used in any form of PH and there should be long-term follow-ups for the thromboembolic type of hypertension.
- Ventilation-perfusion scanning uses medical isotopes to observe air and blood circulation in a patients lungs. It can also observe blood clots if they are present.
- Computed tomography (CT) scans.
- Magnetic Resonance Imaging (MRI).
- Pulmonary angiography uses a dye and X-rays to observe how blood flows through the lungs. This is the standard tool for diagnosis of thromboembolic hypertension. This is often performed in conjunction with right heart catheterization.
Thromboembolic Pulmonary Hypertension Treatment
- Some forms of thromboembolic pulmonary hypertension require surgery, medical treatment or both.
- Endothelin receptor antagonists may be prescribed as they prevent blood vessel constriction. Endothelin is a powerful blood vessel constrictor.
- Endarterectomy is a surgical procedure that unblocks blood vessels. This procedure is done with complete circulatory arrest while the patient is in deep hypothermia between 18°C and 20°C. This procedure is reported to have great success.
- Phosphodiesterase inhibitors are used to allow blood vessels to relax.
- Prostacyclins work well as they relax blood vessels and prevent clot formation.
Thromboembolic Pulmonary Hypertension Prognosis
The mortality rate with individuals diagnosed with thromboembolic pulmonary hypertension is reported to be 90 percent within 3 years after diagnosis.


















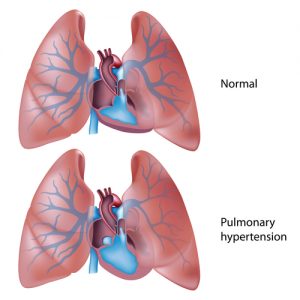


 Tests to confirm PH are important, however, beyond that, patients need to know what caused the onset of pulmonary hypertension. This is important, as it will determine an appropriate treatment(s). Remember: there are many causes for all of the PH forms, so the underlying cause must be determined.
Tests to confirm PH are important, however, beyond that, patients need to know what caused the onset of pulmonary hypertension. This is important, as it will determine an appropriate treatment(s). Remember: there are many causes for all of the PH forms, so the underlying cause must be determined.

